

 Vďaka masívnemu sekvenovaniu DNA sa podarilo objaviť niekoľko stoviek tisíc nových druhov vírusov baktérií – bakteriofágov. O dosiaľ neznámych vírusoch sme sa rozprávali s mikrobiologičkou Máriou Džunkovou.
Vďaka masívnemu sekvenovaniu DNA sa podarilo objaviť niekoľko stoviek tisíc nových druhov vírusov baktérií – bakteriofágov. O dosiaľ neznámych vírusoch sme sa rozprávali s mikrobiologičkou Máriou Džunkovou.
Čo vlastne vieme o bakteriofágoch?
Takmer nič. Veľmi dôležité je dozvedieť sa o nich najmä to, ktorá baktéria je ich hostiteľ. Existujú výpočtové metódy na hľadanie spojitostí medzi fágmi a ich hostiteľskými druhmi baktérií, ktoré sa zakladajú napríklad na hľadaní spoločných sekvencií CRISPR alebo t-RNA. Tieto metódy však fungujú zvyčajne iba pre malé množstvo dobre známych baktérií, no nie pre mnoho novoobjavených bakteriálnych skupín. Teoreticky by sa fágy mohli spojiť s týmito novoobjavenými baktériami pomocou laboratórnych metód, pri ktorých sa analyzujú individuálne bakteriálne bunky pozbierané z environmentálnej vzorky bez nutnosti ich kultivovať, kde by možno náhodou jedna z nich mohla obsahovať vo svojom vnútri nejaký bakteriofág. Takéto metódy sa však nedajú používať na charakterizovanie celého infekčného potenciálu daného fágu.
Ako sa hľadá spojitosť fágov a baktérií?

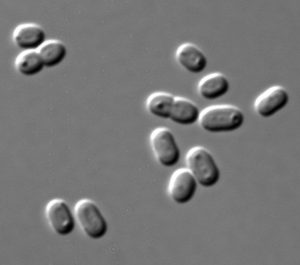
Klasická laboratórna metóda spočíva v tom, že sa k bakteriálnym bunkám rastúcim na Petriho miske pridajú vírusy z environmentálnej vzorky. Ak prebehne infekcia, tak sa na povrchu agaru, prírodnom polysacharide, v ktorom rastú baktérie, objavia priehľadné dierky. To znamená, že sa v nich začal rozmnožovať bakteriofág, ktorý bakteriálne bunky po infekcii roztrhal. Nanešťastie sa táto metóda dá aplikovať iba na baktérie, ktoré sa dajú pestovať v laboratóriu, a je pomalá v porovnaní so stále rastúcim množstvom fágov objaveným vďaka masívnemu sekvenovaniu DNA z environmentálnych vzoriek.
Jestvuje metóda, ktorá by naraz objavila mnoho druhov fágov schopných napadnúť baktériu?
V roku 2014 Li Dengová vyvinula metódu viral tagging, čo by sa dalo preložiť ako značkovanie vírusmi. L. Dengová pestovala oceánsku baktériu Synechococcus, ktorú skombinovala so všetkými fágmi zo vzorky oceánskej vody. DNA týchto fágov zafarbila fluorescentnou zlúčeninou, takže baktérie sa po spojení s fágmi stali tiež fluorescentné. Tieto svietiace baktérie Li vyseparovala pomocou prietokového cytometra, čo je prístroj, ktorý dokáže analyzovať veľkosť a farby tisícok baktérií za sekundu. Pozbieraním svietiacich baktérií získala podskupinu fágov, ktoré boli schopné napadnúť baktériu Synechococcus. Posledný technický krok bolo už len eliminovanie DNA sekvencií baktérie, aby sa ukázalo, ktoré sekvencie patria infekčným fágom. Phil Hugenholtz, austrálsky fylogenetik, chcel túto metódu posunúť ďalej. Je to veľmi potrebné najmä v súčasnosti, keď sa vďaka masívnemu sekvenovaniu DNA denne objavujú nové druhy baktérií, ktoré sa nedajú pestovať v laboratóriu. A tak v rámci nového výskumu zmiešal všetky fágy z jednej environmentálnej vzorky so všetkými baktériami z tej istej alebo inej environmentálnej vzorky.
Aká bola vaša úloha pri tomto výskume?
V Austrálii na mňa čakala ťažká úloha – P. Hugenholtz mi oznámil, že keďže nikto okrem L. Dengovej nebol doteraz schopný viral tagging zopakovať, chce odo mňa nielen to, aby táto metóda fungovala v austrálskom laboratóriu, ale aby som ju vyvinula pre baktérie, ktoré sa nedajú kultivovať.
Podarilo sa to?
Pôvodný viral tagging sa týkal baktérie Synechococcus z morskej vody, ja som chcela pokračovať vo výskume ľudského mikrobiómu zo stolice. To znamená, že aj keď princíp protokolu viral tagging bol pre morskú vodu a ľudský mikrobióm ten istý, musela som všetky podmienky prispôsobiť novému typu vzorky a pracovať s novými kontrolami. Zároveň som všetko robila v mnohých opakovaniach, aby som zistila, ktoré faktory najväčšmi ovplyvňujú úspech experimentu – najmä preto, aby sa neskôr neobjavili pochybnosti, že nikto nevie experiment zopakovať. Najväčší problém bol v tom, že som mala pracovať s baktériami, ktoré sa nedajú kultivovať. V prípade L. Dengovej bol genóm baktérie Synechococcus známy, no v mojom výskume som mala pracovať s baktériami s neznámymi genómami.
Ako ste to riešili?
Keby som pozbierala prietokovým cytometrom všetky baktérie napadnuté fágmi dohromady, ako to bolo s baktériou Synechococcus, nevedela by som rozlíšiť, ktoré sekvencie patria jednotlivým fágom a s ktorými baktériami boli spojené. Preto som zaviedla ukladanie baktérií po jednom do individuálnych jamiek v 384-jamkových doštičkách. Každá individuálna baktéria však obsahuje iba jednu kópiu DNA, čo je málo na to, aby sa dala sekvenovať samostatne. Preto som v prísne sterilných podmienkach použila Phi polymerázu, čo je enzým, ktorý dokáže namnožiť celú molekulu DNA, aby sa dala sekvenovať. Nevýhodou je, že sa niektoré regióny genómu namnožia väčšmi ako iné, a niektoré chýbajú úplne.
Takže ďalší problém…
 Áno, bioinformatický. Zatiaľ čo baktéria má genóm dostatočne dlhý – niekoľko miliónov párov bázy, fágy ho majú dlhý zvyčajne len niekoľko tisíc párov bázy. Identifikácia baktérií je ľahšia aj preto, lebo pre väčšinu z nich máme celkom dobré referenčné genómy. Keďže sú dostatočne dlhé, nejaké menšie či väčšie rozdiely na úrovni izolovaných kmeňov pri identifikácii druhu neprekážajú, dokonca ani chýbajúce časti genómu, čo spôsobila Phi polymeráza. Dokážeme dokonca poskladať genómy úplne neznámych baktérií na základe ich početnosti v environmentálnej vzorke a výskytu kombinácií krátkych DNA sekvencií po celej dĺžke ich genómu.
Áno, bioinformatický. Zatiaľ čo baktéria má genóm dostatočne dlhý – niekoľko miliónov párov bázy, fágy ho majú dlhý zvyčajne len niekoľko tisíc párov bázy. Identifikácia baktérií je ľahšia aj preto, lebo pre väčšinu z nich máme celkom dobré referenčné genómy. Keďže sú dostatočne dlhé, nejaké menšie či väčšie rozdiely na úrovni izolovaných kmeňov pri identifikácii druhu neprekážajú, dokonca ani chýbajúce časti genómu, čo spôsobila Phi polymeráza. Dokážeme dokonca poskladať genómy úplne neznámych baktérií na základe ich početnosti v environmentálnej vzorke a výskytu kombinácií krátkych DNA sekvencií po celej dĺžke ich genómu.
Ako sa klasifikujú novoobjavené baktérie?
Dajú sa klasifikovať pomocou fylogenetických metód. Na rozdiel od baktérií je problém s identifikáciou vírusov v tom, že ich diverzita je obrovská, a teda neexistuje nijaká dosť dobrá databáza referenčných genómov fágov na ich identifikáciu. Genómy fágov sú pomerne krátke a obsahujú gény, ktoré sú spoločné s inými vírusmi. Takže jednoduchým mapovaním sekvencií na referenčné genómy sa nedá dosiahnuť jednoznačný výsledok, najmä keď pracujeme s DNA namnoženou pomocou Phi polymerázy, ktorá nepracuje po celej dĺžke genómu rovnomerne. Pôvodne som si chcela vytvoriť vlastnú databázu vírusov metagenomickým sekvenovaním individuálnych vzoriek stolice do veľkej hĺbky, ale výsledky môjho viral tagging experimentu obsahovali vírusy, ktoré boli pod detekčným limitom metagenomickej analýzy.
Za rozhovor ďakuje redakcia Quarku
Foto archív M. Džunkovej


